



Research Overview:
We use a variety of molecular and bioinformatics approaches to study the evolution of genes, genomes and organisms. A central theme of our research is how genetic interactions (epistasis) and population subdivision change the phenotypic consequences of a gene in different genomic and/or environmental contexts. This research has broad implications for our understanding of topics as seemingly different as mapping genetic causes of disease and the origin of new species.Most of the experimental work in our lab involves flour beetles in the genus Tribolium, but students in the lab are encouraged to pursue questions/systems that appeal to them. Since the growing wealth of genomic data poses exciting opportunities as well as unique challenges, much of our research is integrated with the development of experimental and computational tools necessary to test evolutionary hypotheses on a genomic scale.
Ongoing research topics in the Demuth lab:![]()
Speciation Genetics
If one could conclude as to the nature of the Creator from a study of his creation
it would appear that God has an inordinate fondness for stars and beetles.
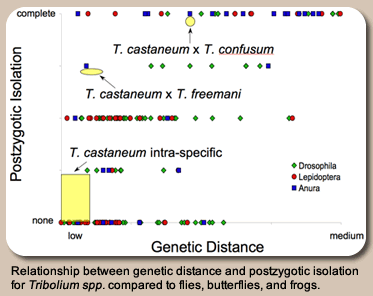 and by the discovery of segregating variation for the ability of different T. castaneum populations to hybridize with their sister species, T. freemani. A long-term research program in the lab seeks to identify the genetic factors acting to isolate populations of T. castaneum from each other and from T. freemani. Ultimately, the goal is to answer general questions about whether the genetic loci isolating populations from each other are of the same type as those separating species (i.e. are they regulatory, coding, or structural?).
We collaborate extensively with Michael Wade's lab at Indiana Univeristy on several of our Tribolium speciation genetics projects.
and by the discovery of segregating variation for the ability of different T. castaneum populations to hybridize with their sister species, T. freemani. A long-term research program in the lab seeks to identify the genetic factors acting to isolate populations of T. castaneum from each other and from T. freemani. Ultimately, the goal is to answer general questions about whether the genetic loci isolating populations from each other are of the same type as those separating species (i.e. are they regulatory, coding, or structural?).
We collaborate extensively with Michael Wade's lab at Indiana Univeristy on several of our Tribolium speciation genetics projects.
Molecular and quantitative genetics of population differentiation:

We initially assayed >200,000 hybrids from ~5,000 families for quantitative genetic analysis and found that epistasis, maternal effects, and genotype-by-environment interactions play important roles in T. castaneum population differentiation. We also found the first instance of environmentally mediated Haldane’s rule, a phenomenon that is typically thought to only occur as a result of fixed differences between species. While we pursue the genetic basis of these findings, we continue to sample populations of Tribolium from disperate locations to find new incompatibilities.
MEDEA: Closer to home, we are also studying the distribution of Medea (Maternal Effect Dominant Embryonic Arrest) in Tribolium populations across Texas. Medea causes a unique phenotype wherein offspring of Medea/+  mothers who do not inherit their mother's Medea factor all die. This factor has become increasingly interesting to us since, 1) it acts like a "speciation gene" when combined with a second factor found in T. castaneum populations from India; and 2) Lorenzen et al 2008 mapped one Medea factor (M1) to the insertion of a large transposon insertion, and studying the biology of transposable element movement is a major topic of research in the Feschotte, Pritham, and Christensen labs here at UTA
mothers who do not inherit their mother's Medea factor all die. This factor has become increasingly interesting to us since, 1) it acts like a "speciation gene" when combined with a second factor found in T. castaneum populations from India; and 2) Lorenzen et al 2008 mapped one Medea factor (M1) to the insertion of a large transposon insertion, and studying the biology of transposable element movement is a major topic of research in the Feschotte, Pritham, and Christensen labs here at UTA
Mapping incompatibility factors:
Because mapping traits that have complex genetic architectures is notoriously difficult, our mapping efforts involve development of recombinant congenic strains (RCS). RCS isolate genes that cause negative consequences in a benign genomic context and can then be "tested" against an incompatible genome. These lines will become a community resource that may be useful for dissecting the genetic basis of many phenotypes. In convert with the wealth of information generated my the T. castaneum genome sequence the potential exists for  very high resolution mapping.
very high resolution mapping.
Genetic basis of Haldane's Rule between Silene species:
We have an ongoing collaboration with Lynda Delph's lab at Indiana University to study the quantitative genetics of reproductive isolation in a group of plants that has both hermaphroditic and dioecious species. Her studies may provide the first evidence for Haldane's Rule in a plant. We are helping to design crosses and tease appart the quantitative genetic basis for hybrid breakdown.
![]()
Computational Genomics
There is a growing appreciation that many major morphological and functional changes in evolution result from duplications that range in size from genes to whole genomes. Furthermore, differential silencing of duplicate gene copies between populations may contribute to speciation. Unlike the analysis of orthologous sequences, where there are widely accepted neutral expectations for molecular evolution, there is no corresponding framework for the study of gene duplication and loss. While many studies attribute large changes in the size of gene families to natural selection, there is no solid theoretical foundation for what might be expected purely due to stochastic gain and loss of genes. To address this deficiency, we have developed a likelihood-based method that makes efficient use of genomic data in a phylogenetic context. The method allows us to identify gene families that are expanding or contracting significantly faster than can be explained by a random process. The Computational Analysis of Gene Families (CAFE) software can be downloaded from Matthew Hahn's lab website here.Gene family evolution in mammals:
Studying gene duplication and loss from a comparative genomics perspective requires related taxa with excellent sequencing coverage so that a minimal number of duplicates are missing, or collapsed, in the genome assemblies. Among eukaryotes, mammals currently have the best-sequenced group of taxa for genome-scale analysis of gene family evolution. Therefore, as part of the macaque genome project, we used CAFE to analyze gene family evolution across the genomes of human, chimpanzee, macaque, mouse, rat, and dog.
evolution. Therefore, as part of the macaque genome project, we used CAFE to analyze gene family evolution across the genomes of human, chimpanzee, macaque, mouse, rat, and dog.
Results from our analysis of mammalian gene family evolution contribute to answering the long-standing paradox of how humans and chimpanzees can be so similar at the nucleotide level yet so phenotypically different. Briefly, we found that in addition to the regulatory and coding sequence changes often cited as being only ~1.5% divergent, there are a large number of differences in gene complement between species that are necessarily unaccounted for in comparisons of orthologous sequences. Additionally, while all of the genomes in our analysis contain some families that are changing faster than the random rate, there has been a significant acceleration in the average rate of gene gain and loss in humans and chimpanzees compared to macaque and the other mammals. Pursuing the mechanisms responsible for this overall acceleration as well as the families that have undergone significant changes between humans and chimpanzees is an exciting area of ongoing research. In the near future we expet to have complete genomes for several Tribolium species, allowing us to conduct gene family analysis in that system.
Molecular evolution of duplicated genes:
In addition to understanding the extent to which gene duplication and loss have contributed to genome evolution, we are studying the mechanisms responsible for the evolution of individual duplicates. New gene duplicates ultimately suffer one of three fates: 1) one copy may evolve  new function, 2) the two copies may partition ancestral function, or 3) one copy may become lost by deletion or pseudogenization. Integrating molecular population genetics with comparative approaches, we have shown that when one copy of the duplicates undergoes a regulatory change resulting in sex-limited gene expression, selection is relaxed. In at least one case, this relaxed constraint appears to have contributed to the evolution of novel function.
new function, 2) the two copies may partition ancestral function, or 3) one copy may become lost by deletion or pseudogenization. Integrating molecular population genetics with comparative approaches, we have shown that when one copy of the duplicates undergoes a regulatory change resulting in sex-limited gene expression, selection is relaxed. In at least one case, this relaxed constraint appears to have contributed to the evolution of novel function.
While my empirical studies in this area have thus far focused on sequencing Drosophila developmental genes (bicoid and zen), the potential for genes with sex-limited expression to evolve rapidly may have important implication for Tribolium speciation. The quantitative genetic studies show that there is a strong maternal effect component to population differentiation. We are currently identifying potential maternal effect genes in the Tribolium genome using custom microarrays (see below).
![]()
Tribolium Genomics
We have developed a Roche/NimbleGen 384k microarray to simultaneously assess gene expression for all of the predicted genes in the T. castaneum genome. Our aim is to use these arrays to elucidate missexpressed genes in inter-population hybrids by comparing expression to offspring of intra-population matings. To date we have collected baseline data for several developmental time points and tissues in males and females.
Sex-biased expression and dosage compensation:
A genome's ability to produce two separate, sexually dimorphic phenotypes is an intriguing
biological mystery. Microarray-based studies of a handful of model systems suggest that much of
the mystery can be explained by sex-biased gene expression evolved in response to sexually
antagonistic selection. We present the first whole genome study of sex-biased expression in the
red flour beetle, Tribolium castaneum. Tribolium is a model for the largest eukaryotic order,
Coleoptera, and we show that in whole body adults, ~20% of the transcriptome is differentially
regulated between the sexes. Among T. castaneum, Drosophila melanogaster, and Anopheles
gambiae we identify 416 1:1:1 orthologs with conserved sex-biased expression.  Over
represented functional categories among sex-biased genes are primarily those involved in gamete
production and development. Most importantly, the genomic distribution of sex-biased genes in T. castaneum is distinctly non-random, with the strongest deficit of male-biased genes on the X
chromosome (9 of 793) of any species studied to date. Tribolium also shows an enrichment of X
linked female-biased genes (408 of 793). The extensive feminization of Tribolium X-chromosome gene expression appears to be due to unmitigated hyperexpression of X-linked
genes in both sexes. We propose that the over-expression of X-chromosomes in females is an
evolutionary side-effect of the need to dosage compensate in males, and that the current status
may reflect a stable equilibrium wherein females are sufficiently, though not completely, dosage
compensated.
Over
represented functional categories among sex-biased genes are primarily those involved in gamete
production and development. Most importantly, the genomic distribution of sex-biased genes in T. castaneum is distinctly non-random, with the strongest deficit of male-biased genes on the X
chromosome (9 of 793) of any species studied to date. Tribolium also shows an enrichment of X
linked female-biased genes (408 of 793). The extensive feminization of Tribolium X-chromosome gene expression appears to be due to unmitigated hyperexpression of X-linked
genes in both sexes. We propose that the over-expression of X-chromosomes in females is an
evolutionary side-effect of the need to dosage compensate in males, and that the current status
may reflect a stable equilibrium wherein females are sufficiently, though not completely, dosage
compensated.
Tribolium confusum genome sequencing:
T. confusum is morphologically and behaviorally very similar to T. castanuem but has one fewer chromsome due to an X - Autosome fusion. In conjunction with my Spring 2010 Bioinformatics course we are sequencing the Tribolium confusum genome on our new 454 Genome Sequencer. Some of the interesting things we hope to learn include: 1) what happens to the composition and evolutionary rate of genes on the neo-X in T. confusum, 2) how does the transposon compliment differ between T. castaneum and T. confusum, and 3) how do some of the large gene families discovered in T. castaneum (e.g. P450s and odorant receptors) differ in T. confusum?
![]()
Sexual Selection in Horned Beetles
The broad horned flour beetle (Gnathocerus cornutus) has a pronounced sexual dimorphism wherein males have large mandibular horns but females do not. Males use these horns in physical combat to gain access to mates. There are two general hypotheses for the evolution of exaggerated male (or female) traits that arise as a consequence of sexual selection. First, the handicap hypothesis posits a conflict between sexual selection and natural selection where sexually selected traits benefit an individual’s probability of successful mating at the expense of other fitness aspects (e.g. survival). The alternative hypothesis is that exaggerated traits serve as an  indicator of genetic superiority. This so called "good-genes" hypothesis suggests, for example, that the offspring of a female who mates with a showy male will accrue an indirect benefit by inheriting his superior genes. Amrita Naidu recently completed her M.S. degree in the lab studying the effect of parasite infection on horn size and mating success. Her principle finding was that infection significantly decreases horn size and that horn size is the primary determinant of mating success. Amrita looked at immune protein concentrations in infected beetles but she was unable to find an increased immune response in infected beetles. This is puzzling given that beetles were clearly infected (parasites observed via dissection). Currently no one in the lab has taken up this system, but it presents a number of exciting possibilities to follow up on Amrita's thesis work, particularly since many of the Tribolium genomic tools may be easily transferred to Gnathocerus.
indicator of genetic superiority. This so called "good-genes" hypothesis suggests, for example, that the offspring of a female who mates with a showy male will accrue an indirect benefit by inheriting his superior genes. Amrita Naidu recently completed her M.S. degree in the lab studying the effect of parasite infection on horn size and mating success. Her principle finding was that infection significantly decreases horn size and that horn size is the primary determinant of mating success. Amrita looked at immune protein concentrations in infected beetles but she was unable to find an increased immune response in infected beetles. This is puzzling given that beetles were clearly infected (parasites observed via dissection). Currently no one in the lab has taken up this system, but it presents a number of exciting possibilities to follow up on Amrita's thesis work, particularly since many of the Tribolium genomic tools may be easily transferred to Gnathocerus.
